Case 54
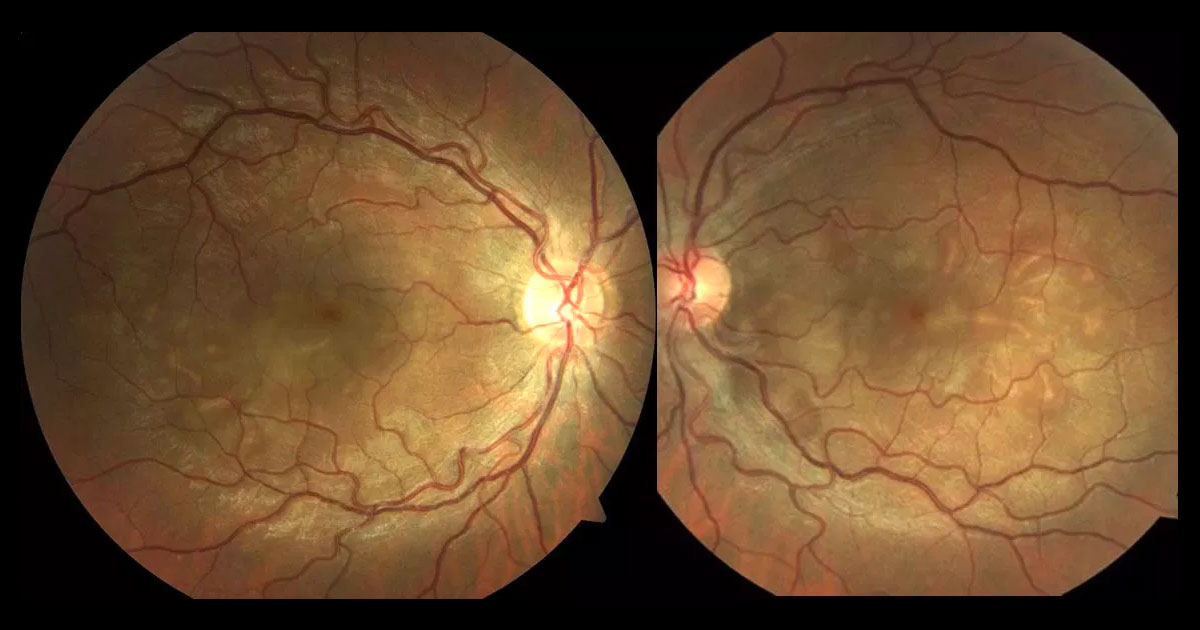
Figure 1. Colour fundus photography shows multiple, bilateral serous retinal detachments. The discs are normal and there is no retinal vasculitis.
Author: Simon Nothling Editor: Adrian Fung
A 36-year-old Caucasian male was referred with headache, orbital pain, vertigo and blurred vision.
Case history
A 36-year-old Caucasian male, presented with a 2-day history of bilateral blurred vision, which was preceded by a headache, orbital pain and vertigo for 3 days. He had presented to an emergency department 2-days prior where he had been diagnosed with a viral infection and dehydration before being discharged home. His wife was 37 weeks pregnant, and he described recent sleep loss and stress. He seen by an optometrist who noticed macula swelling and referred the patient to our clinic. His main visual symptoms were distortion and central blurring. He had no other neurological symptoms such as tinnitus, paraesthesias, diplopia or speech disturbance. He was otherwise healthy with no other medical conditions, and took no regular medication. There was no history of ocular surgery or trauma.
Visual acuities were 6/15 in the right eye (OD) and 6/6 in the left eye (OS). Intraocular pressures were 15mmHg in each eye. Pupils were normal with no RAPD. Anterior segment exam was normal, with no anterior chamber cells, and no cells in the anterior vitreous. Dilated fundus photos of both eyes showed bilateral, multiple, blisters of subretinal fluid (Figure 1).
What is your diagnosis?
Click to reveal answer
Differential diagnosis
The differential diagnosis of serous retinal detachment includes:
- Central Serous Chorioretinopathy
- Vogt-Koyanagi-Harada Disease
- Sympathetic ophthalmia
- Syphilis chorioretinitis
- APMPPE
- Tuberculosis chorioretinitis
- Sarcoidosis
- Lupus choroidopathy
- Malignant hypertension
- Posterior scleritis
Additional history, examination and investigations
Blood pressure was 130/80mmHg. Optical coherence tomography demonstrated multiple blisters of subretinal and intraretinal fluid in both eyes with thickened choroids (Figure 2). The areas of subretinal fluid corresponded with areas of hyperautofluorescence (Figure 3). Fluorescein angiography showed early hypofluorescent spots and later hyperfluorescent dots, with hyperfluorescent pooling within the areas of serous retinal detachment (Figure 4). Indocynaine green angiography (ICG) showed multiple hypofluorescent spots (Figure 5). B-scan ultrasonography demonstrated a thickened choroid (Figure 6).
Blood tests were all normal, including: FBC, UEC, ESR, CRP, syphilis serology, Quantiferon-gold, ACE, ANA, ANCA and anti-ds-DNA. Chest X-ray was normal.

Figure 2. Optical coherence tomography shows multiple bilateral exudative retinal detachments, outer retinal cysts and thickened choroids.

Figure 3. Fundus autofluorescence shows hyperfluorescsent areas corresponding to the subretinal fluid.

Figure 4. Fluorescein angiography demonstrates hypo fluorescent spots in the early phases A), with pin-point hyperfluorescence and hyperfluorescent pooling in the later phases B).

Figure 5. Indocyanine green angiography shows multiple hypofluorescent spots.

Figure 6. B-scan ultrasound shows a thickened choroid , but no evidence of posterior scleritis.
DIAGNOSIS
The patient was diagnosed with Vogt-Koyanagi-Harada disease (VKH).
Clinical course
With such severe macula abnormalities on OCT, and symptomatic vision loss, the patient was commenced on high dose oral prednisolone. Infective causes were considered unlikely on history due to a lack of risk factors, and serological blood tests excluded tuberculosis and syphilis. Prednisone 100mg daily was initially commenced with omeprazole 20mg (peptic ulcer prophylaxis) and calcium and vitamin D supplementation.
Three days later the patient reported an improvement in vision with resolution of his headache and vertigo. Visual acuities had improved to 6/7.5 OD and 6/6 OS. Progression OCT scans over 10 days demonstrated rapid resolution of subretinal and intraretinal fluid with thinning of the choroid (Figure 7). The patient was reviewed closely and by 8 weeks the prednisone had been tapered to 20mg/day with normalisation of OCT scans. No skin, hair or hearing problems have been identified.

Figure 7. Progression OCT scans over 10 days shows reduction of subretinal fluid, intraretinal fluid and thinning of the choroids
Discussion
The diagnosis of VKH can be a challenging, particularly with its varied presentation, at various clinical stages. The combination of neurological symptoms and bilateral exudative retinal detachments (subretinal fluid) should raise high suspicion for the acute uveitic stage of VKH.
Unlike our patient, VKH is most commonly seen in pigmented races, such as Asians, Hispanics, American Indians, and Asian Indians. It appears to be more common in Japan, where it accounts for 6.7% of all uveitis referrals. In the USA, it accounts for 1-4% of all uveitis clinic referrals. Most studies report women to be more affected than men, and most patients are 20-40 years of age.(1)
The primary pathology appears to be an autoimmune condition against melanocyte containing tissues such as uvea, ear, meninges and skin.(2) This explains the additional extraocular features of the disease, such as vitiligo, poliosis, tinnitus, vertigo and alopecia. Many of the skin findings are not found until the later stages of the disease.
In 2001, the First International Workshop on Vogt-Koyanagi Harada Disease revised the diagnostic criteria for VKH.(3) The criteria are as follows:
- No history of penetrating trauma or surgery
- No clinical or laboratory evidence suggestive of other ocular disease
- Bilateral ocular involvement (early disease: focal areas of subretinal fluid or bullous serous retinal detachments, choroidal thickening and fluorescein angiography changes such as multifocal areas of pinpoint leakage. Late disease: sunset glow fundus, retinal pigment epithelial clumping and migration)
- Neurological/auditory findings at any point during the course of the disease (eg. meningismus symptoms such as headache, fever, malaise, or stiff neck, tinnitus, or CSF fluid pleocytosis)
- Integumentary findings not preceding the onset of other central nervous system disease (eg. alopecia, vitiligo, poliosis)
There are 4 stages to VKH. First is the prodromal stage, which may only last a few days, and can present similar to a viral-like illness. Headaches, nausea, dizziness, fever, neck stiffness and focal neurological signs can occur. The acute uveitic stage presents with a bilateral posterior uveitis, in the form of a multiple serous retinal detachments, and hyperaemia of the optic nerve heads. This is the stage our patient presented with. The OCT, FFA and ICG are all key investigations at this stage. The OCT shows pockets of subretinal fluid, often “tacked down” to the retinal pigment epithelium, presumably by fibrin.(4) Pigment epithelial detachments are absent. In 40% of cases there are intraretinal cysts, which differs from most other causes of cystoid macular oedema by its predominance in the outer retina with preservation of the inner retina. Enhanced-depth imaging OCT will often show thickening of the choroid, which can be used to monitor disease activity. Fluorescein angiography typically shows pin-point hyperfluorescence in a “starry sky” appearance. The third stage is the chronic uveitic stage which is characterised by the development of vitiligo, poliosis and depigmentation of the choroid (“sunset-glow fundus”). The fourth stage is the chronic recurrent stage where episodes of granulomatous anterior uveitis occur, and vision-threatening complications such as subretinal neovascular membranes, glaucoma and cataract develop.
The differential diagnosis includes sympathetic ophthalmia, hence the need to exclude prior trauma or surgery. Central serous chorioretinopathy can present with serous retina detachments and a thickened choroid, but it may also have pigment epithelial detachments, the optic discs are not hyperemic and there are no systemic symptoms. It is important to exclude other infectious and inflammatory conditions.
Vogt-Koyanagi-Harada disease disease needs early, aggressive treatment with high dose corticosteroids to shorten disease duration and to reduce the risk of vision threatening complications and extraocular manifestations developing. Prednisone is normally started in 80-100mg orally (or 1-2mg/kg) with an individualised taper over a 6 month period.(5) 60% of patients achieved better than 6/12 vision with this treatment regimen.5 Recurrences of the disease are not so steroid responsive, and may require other immunosuppressive agents such as azathioprine, methotrexate, mycophenolate, or cyclosporine.
TAKE HOME POINTS
- The acute uveitic stage of VKH can present with neurological symptoms (eg. headache, neck stiffness, tinnitus) and a rapid deterioration in vision in both eyes from bilateral multifocal exudative retinal detachments. The optic discs are often hyperaemic.
- OCT scans show pockets of subretinal fluid, outer-retinal cystic oedema, a thickened choroid and absence of pigment epithelial detachments.
- Fluorescein angiography classically shows pin-point hyperflourescence lesions, and dye pooling into the subretinal space
- Early treatment with high-dose corticosteroids leads to an improved visual outcome and less risk of recurrence.
REFERENCES