Case 21
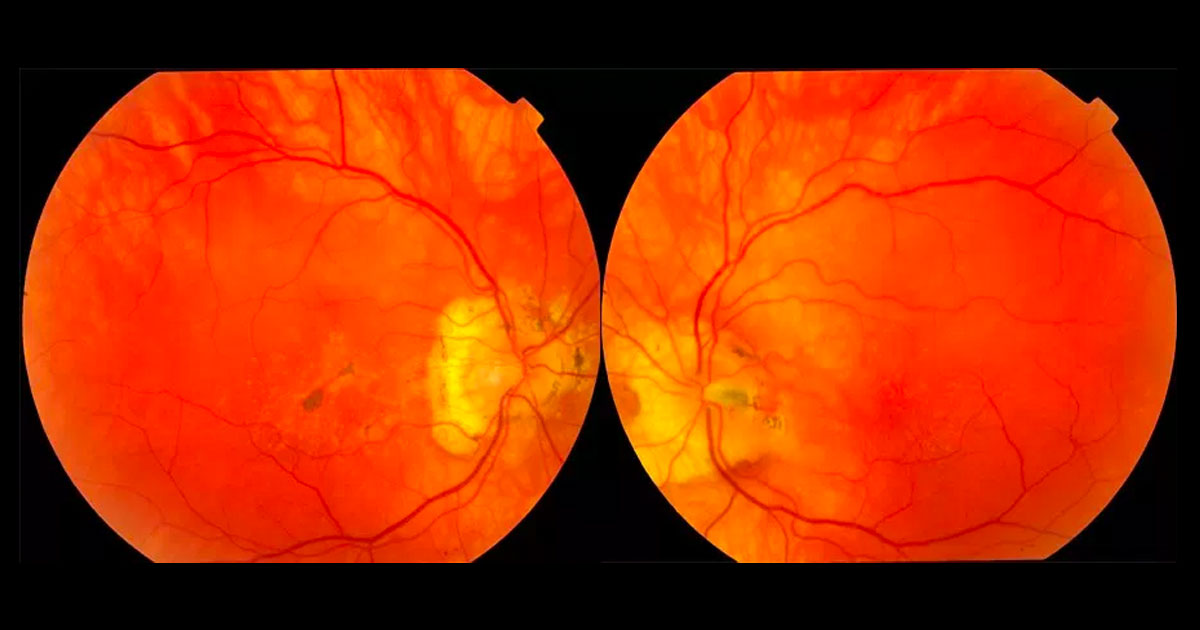
Figure 1. Colour fundus photographs showing multiple, well-circumscribed deep drusenoid deposits in the maculae and peripapillary regions. There are associated pigmentary changes in the right macula and an incidental left optic disc pit.
Author: Raj Chalasani Editor: Adrian Fung
A 41-year-old male was referred with distortion in his right eye.
Case history
A 41-year-old male presented with a 6-week history of distortion in his right eye. He was otherwise systemically well. He was born in England and had immigrated to Australia at the age of 15 years.
Best corrected visual acuities were 6/7.5 in the right eye (OD) and 6/6 in the left eye (OS). Examination of the fundi revealed multiple, well-circumscribed, yellow and deep drusenoid deposits in the macula and nasal peripapillary region of both eyes (Figure 1). Pigmentary changes were noted in the right macula. The left optic nerve showed an incidental finding of an optic disc pit.
What is your diagnosis?
Click to reveal answer
Differential diagnosis
The differential diagnosis of bilateral, multiple, yellow and deep macular drusenoid deposits includes:
- Age-related macular degeneration
- RDS/Periphern gene mutations (Pattern Dystrophies)
- ABCA4 gene mutations (Stargardt Disease, Fundus Flavimaculatus)
- Familial Dominant Drusen (Malattia Leventinese, Doyne Honeycomb Retinal Dystrophy)
- Sorsby Macular Dystrophy
Additional history, examination and investigations
The patient reported that his father and sister had both been diagnosed with ‘macular degeneration’ at an early age.
Optical Coherence Tomography (OCT) of the right eye localises the deposits to the sub-retinal pigement epithelial (RPE) region, confirming the lesions to be drusen (Figure 2). Red-free photography demonstrates a radiating pattern of drusen within the macula and peripapillary regions of both eyes (Figure 3). Autofluorescence is altered in the area of macula drusen and peripapillary regions of both eyes (Figure 4). Genetic testing was offered, but the patient declined.

Figure 2. Optical coherence tomography localises the lesions to a sub-RPE location.

Figure 3. Red-free photography demonstrates the radial pattern of the drusen.

Figure 4. Autofluorescence is altered in the area of macula drusen and peripapillary regions of both eyes.
DIAGNOSIS
Presumed Familial Dominant Drusen (Malattia Leventinese).
Clinical course
The patient is currently under intermittent observation and the vision and symptoms have remained stable.
Discussion
Although a definitive diagnosis cannot be made without genetic confirmation, the presumed diagnosis is Malattia Leventinese. Malattia Leventinese is a rare form of familial drusen that shows autosomal dominant inheritance.(1,2) It is so named due to a number of individuals from a family in the Leventine valley of southern Switzerland who were found to have the condition in the 1920’s (“Malady of the Leventine Valley”). There are strong similarities with Doyne Honeycomb Retinal Dystrophy (described in a group of English patients in 1899), due to involvement of similar regions of chromosome 2p.(1-3)
The key clinical findings are radiating, elongated drusen visible at the maculae from the second decade of life, often in association with nasally located drusen, which are rare in other types of macular degeneration.(4)
Patients typically remain asymptomatic until the 4th decade, when reduced vision or metamorphopsia may prompt presentation. Atrophy or choroidal neovascularisation (CNV) leads to further visual loss as the patient ages.
No treatment is currently available unless CNV develops, in which case intravitreal anti-VEGF agents can be utilised. Genetic testing is available (mutations may be found in the EFEMP1 gene(3)), and genetic counselling is beneficial for patients planning on having a family.
TAKE HOME POINTS
- Drusenoid deposits are not always associated with Age-related Macular Degeneration. Consider other diagnoses, especially if the patient is under 50 years of age.
- Malattia Leventinese is a rare form of familial drusen characterised by radiating drusen in the macula and peripapillary regions. The inheritance is autosomal dominant and there is typically a history of affected family members.
- Patients are usually asymptomatic until middle-age unless choroidal neovascularisaton develops.
- Genetic testing and counselling is available.
REFERENCES