Case 29
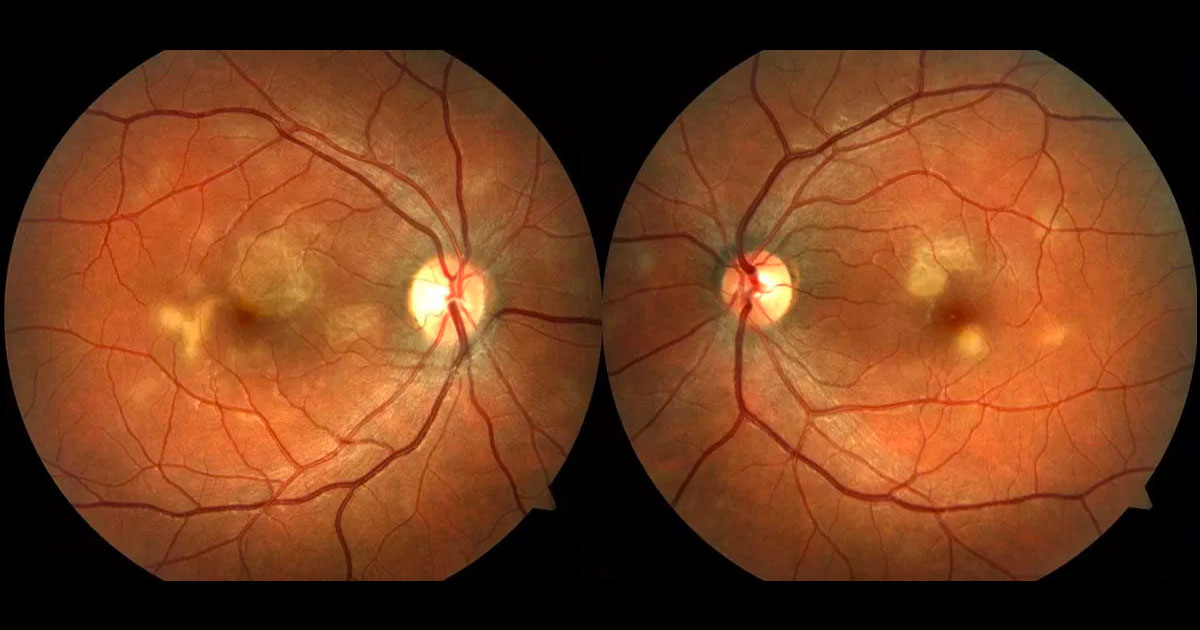
Figure 1. Colour fundus photographs showing bilateral multiple cream coloured deep retinal placoid lesions. The right fovea is involved.
Author: Simon Nothling Editor: Adrian Fung
A 24-year-old female engineer presented with a 1 week history of bilateral blurred vision with paracentral scotomas and occasional flashes.
Case history
A 24-year-old female engineer presented with a 1 week history of bilateral blurred vision with paracentral scotomas and occasional photopsiae. She was a low myope, but had no other ophthalmic history. Two months prior she had suffered from a severe viral upper respiratory tract infection. There were no neurological symptoms such as limb weakness, speech disturbance or headaches. She had no past medical history, and was not on regular medication. There was no recent overseas travel.
On examination, best corrected visual acuities were 6/7.5-2 right eye (OD) and 6/5 left eye (OS). Intraocular pressures were 8mmHg (OD) and 9mmHg (OS). There was 1+ white blood cells in both anterior chambers, but no vitritis. Dilated fundoscopy showed multiple cream coloured deep retinal placoid lesions involving both maculae (Figure 1). The right macula had larger lesions, with the superior lesion involving the fovea. The lesions appeared to be of different ages as some had scattered pigmentary changes. There was no disc swelling or vasculitis in either eye.
What is your diagnosis?
Click to reveal answer
Differential diagnosis
- Acute Posterior Multiple Placoid Pigment Epitheliopathy (APMPPE)
- Punctate Inner Choroidopathy (PIC)
- Macular Serpiginous Choroidopathy
- Multiple Evanescent White Dot Syndrome (MEWDS)
- Sarcoidosis
- Tuberculosis
- Syphilis
- Choroidal metastases
Additional history, examination and investigations
Fundus autoflourescence showed the lesions to be hypoautoflourescent centrally, with some hyperautoflourescent changes more at the periphery of the lesions (Figure 2). Fluorescein angiography showed early hypoflouresence of the lesions but late hyperflourescence (Figure 3). The lesions were hypoflourescencent in in the early and late films of indocyanine green angiography, and much more extensive than the lesions seen clinically (Figure 4). Optical coherence tomography (OCT) showed hyper-reflectance throughout the outer nuclear layer, corresponding to the placoid lesions (Figure 5). In these areas, there was also disruption of the ellipsoid line (inner segment/outer segment junction), suggesting injury to the photoreceptors. The right fovea was more affected than left, which corresponds to the worse vision in the right eye.
Blood tests results showed a normal full blood count, urea/creatinine electrolyes and serum ACE. Quantiferon-gold, syphilis serology, ANA and ANCA were all negative. Chest x-ray was normal.

Figure 2. On fundus autofluorscence the lesions are centrally hypoautofluorescent with some hyperautofluorescent changes at the edges.

Figure 3. On fluorescein angiography the lesions are hypofluorescent early but hyperfluorescent in the late phases.

Figure 4. The lesions are hypofluorescent on indocyanine green angiography. They are much more extensive than seen on clinical examination (compare with Figure 1).

Figure 5. Optical coherence tomography shows the placoid lesions to be hyper-reflectivities in the outer nuclear layer, with disruption of the photoreceptors.
DIAGNOSIS
A diagnosis of Acute Posterior Multiple Placoid Pigment Epitheliopathy (APMPPE) was made, supported by the fluorescein angiography which showed classic signs of early hypoflourescence and late hyperflourescence of the lesions.
Clinical course
Once infective causes, such as tuberculsois and syphilis, had been excluded, the treatment option of oral steroids were discussed. It was explained to the patient that most cases of APMPPE do not require treatment, and improve spontaneously over 4-6 weeks. However, as the placoid lesions in her right eye were involving the fovea and the patient was keen for treatment, a short course of oral prednisone was prescribed. She was consented for side-effects of systemic steroids including avascular necrosis of the head of femur and immunosuppression and had no contra-indications such as peptic ulcer disease or diabetes.
Oral prednisone 40mg daily (she weighed 50kg) was commenced, with a 4 week taper. She was also commenced on omeprazole 20mg daily to protect her from developing a gastric ulcer. When she was reviewed in 2 weeks, her vision had improved to 6/5 OD, 6/5. Subjectively, she had also noticed her vision improving with less difficulties at work reading the computer screen. She completed her course of oral prednisone without any side effects.
Over the next 3 months the pale lesions began to fade, and were replaced by some pigmentary changes (Figure 6). Vision remained 6/5 OU. The OCT appearance of both eyes also showed improvements with a reduction in size of the hyperreflective outer nuclear lesions and less disruption of the elipsoid lines (Figure 7). Humphrey Visual Fields also showed improvement (Figure 8).

Figure 6. Three months after initial presentation the lesions are less prominent and have started to pigment.

Figure 7. Three months after initial presentation the lesions are less prominent on optical coherence tomography.

Figure 8. Humphrey visual fields at presentation (top images) and 3 months later (bottom images). The bilateral central scotomas have almost resolved during this time.
Discussion
Acute posterior mulitifocal placoid pigment epitheliopathy (APMPPE) was described by Donald Gass in 1968,(1) and is characterised by a rapid, transient loss of visual acuity associated with an acute onset of multiple, discrete yellow-white placoid lesions at the level of the retinal pigment epithelium (RPE) in the posterior pole. At least 75% of cases are bilateral,(2) and males are equally as affected as females. About 1/3 of cases are preceded by a viral illness. Other visual symptoms include paracentral scotomas, metamorphopsia, “spots” in the vision, and photopsies. Typically this condition presents between the ages of 20 and 50 years, with the mean age of onset being 26 years (similar to our patient). Vitritis is not a significant component of APMPPE, but patients can even present with an anterior uveitis, such as in this case. Other inflammatory “white dot” syndromes often present with significant vitritis, such as birdshot chorioretinopathy and multifocal choroiditis with panuveitis, so this can be a useful differentiating factor.
Most patients with APMPPE demonstrate spontaneous improvement over 2-4 weeks. However, 25% of patients still end up with a visual acuity of 6/12 or worse, so the prognosis is not as good as once thought.(2) Most patients who do not achieve a complete recovery have foveal involvement at presentation, so there is a trend to treat these patients with corticosteroids. Those without foveal involvement normally make a good recovery without any treatment. There are no scientific studies proving that steroids are useful to treat APMPPE, but in more severe cases, they should be considered. As with any cause of uveitis, it is important to exclude infections such as syphilis and tuberculsosis, before commencing steroids. It is imperative that APMPPE patients are asked about neurological symptoms such as severe headache, limb weakness, paraesthesias or disturbance, in case a cerebral vasculitis is present. These patients need an urgent cerebral magnetic resonance angiography (MRA), and if cerebral vasculitis is present, they urgent ueurological admission for intravenous steroids(3).
Additional ophthalmic investigations certainly assist in making the diagnosis of APMPPE and differenting this from other “white dot” syndromes. Gass(1) described the lesions in early phase fluorescein angiography as non-fluorescent, with obstruction of choroidal fluorescence. Later in the angiogram there is a progressive, irregular staining of the lesions, thus giving the usual finding of “early hypoflourescence and late hyperflourescence”. Indocyanine green angiography (ICG) shows hypoflourescence throughout the entire study, early and late, which gave way to the theory that APMPPE is due to choriocapillaris occlusion. The exact pathogenesis of the disease is still debated today, as Gass believed the primary pathology was at the level of RPE(1). Fundus autofluorescence in acute APMPPE shows the lesions to be hypoautoflourescent in the centre, with some hyperautoflourescence at the lesion borders.(4) During the recovery phase, the hyperautoflourescence areas fade, indicating recovery of the RPE. The OCT changes in the acute setting of the placoid lesions show hyperreflectivity changes in the outer retinal layers with disruption of the phtoreceptors.(5) As the vision recovers, there is a recovery of the photoreceptors, but this is not always complete, which is why some patients with APMPPE are left with some permanent visual disability.
TAKE HOME POINTS
- APMPPE is a rare “white dot syndrome” characterised by bilateral, deep creamy white lesions with the posterior poles.
- Male and females are equally affected, and it usually affects patients of young to middle-age.
- Symptoms include blurred vision, central scotomas and photopsiae.
- Fluorescein angiography typically shows early hypofluorescence and late hyperfluorescence of the lesions.
- Oral steroids may need to be considered if the fovea is involved/threatened or there are signs of cerebral vasculitis.
- Visual prognosis is relatively good, but some patients make an incomplete recovery.
REFERENCES